Adrian M. Senderowicz; Information Needed to Conduct First-in-Human Oncology Trials in the United States: A View from a Former FDA Medical Reviewer. Clin Cancer Res 15 March 2010; 16 (6): 1719–1725. https://doi.org/10.1158/1078-0432.CCR-09-2766
Download citation file:
toolbar searchAny drug product not previously authorized for marketing in the United States requires the submission of an Investigational New Drug application (IND). Although the IND submission is regulated by law (21CFR 312), there are several issues that are not covered in the law or U.S. Food and Drug Administration (FDA) guidances that are important for a successful IND submission. For oncology products, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S9 guidance (still in draft) is the most relevant. The most difficult issues to solve in an IND are chemistry, manufacturing and control information, and pharmacology and toxicology. In the United States, pivotal toxicological studies are done in two species: one rodent (i.e., rats) and one nonrodent (i.e., dogs). The safe starting dose is based on toxicological findings observed in the most sensitive species. Most first-in-humans studies in oncology include patients with advanced and/or metastatic disease, as serious to severe side effects of anticancer therapies are often less threatening to advanced cancer patients than their disease, and acceptable levels of toxicity are higher. For other indications (adjuvant therapy, chemoprevention, or healthy volunteers), first-in-human studies need to follow ICH M3 guidelines as the risk to benefit ratio in those subjects and/or patients without evidence of tumor is different. The division welcomes submissions before the IND, also known as pre-INDs, particularly for products with “atypical issues.” Clin Cancer Res; 16(6); 1719–25
This issue of CCR Focus is aimed at understanding the complex issues that are involved in the conduct of phase I trials. Several innovative phase I trial designs have been proposed but in general not often implemented, despite the fact that the designs attempt to reduce the number of patients receiving subtherapeutic doses and increase the efficiency of trials (1, 2). Going forward, the more frequent incorporation of biomarkers has the possibility to aid in patient selection, again potentially improving efficiency (3). Ultimately, however, the outcome of a phase I trials rests, at least in part, upon the quality of preclinical work that was carried out to support the Investigational New Drug (IND) submission. This topic is the scope of this article.
As clearly stated in the U.S. regulations [Title 21 Code Federal Regulations (CFR)], “Under current regulations, any use in the US of a drug product not previously authorized for marketing in the US first requires submission of an IND to the FDA” (21 CFR 312.22 and 312.23). Thus, all new drugs need to have an IND before testing in humans. However, in order to successfully submit an IND, specific relevant information needs to be included. To this end, several guidances related to IND submissions are available (see Table 1). The two most relevant guidances for oncology products are S9 and M3. The sections needed for a new IND are described in 21 CFR 312.23 (Table 2).
FDA guidance relevant for IND submission
| S1 Carcinogenicity, |
| S2 Genetic toxicity, |
| S3 Toxicokinetics, |
| S4 Duration of Chronic Toxicity Testing, |
| S5 Reproductive toxicity, |
| S6 Biotechnology, |
| S7 Safety Pharmacology, |
| S9 Nonclinical Studies for Development Anticancer Drugs and Biologics (under review), |
| M3 Nonclinical Safety Studies for the Conduct of Human Clinical Trials. |
| S1 Carcinogenicity, |
| S2 Genetic toxicity, |
| S3 Toxicokinetics, |
| S4 Duration of Chronic Toxicity Testing, |
| S5 Reproductive toxicity, |
| S6 Biotechnology, |
| S7 Safety Pharmacology, |
| S9 Nonclinical Studies for Development Anticancer Drugs and Biologics (under review), |
| M3 Nonclinical Safety Studies for the Conduct of Human Clinical Trials. |
NOTE: From the FDA Guidance (Drugs) website (23).
Elements of an IND
| Cover sheet: FDA form 1571 |
| Table of contents |
| Introductory statements and general investigations: developmental plan for the drug into perspective and plans contingent on the outcome of the studies |
| Investigator's brochure |
| Chemistry, manufacturing, and control information |
| Pharmacology and toxicology information |
| Protocols (21 CFR 312.23(a) (6) |
| Previous human experience with the investigational drug: presented in an integrated summary report |
| Cover sheet: FDA form 1571 |
| Table of contents |
| Introductory statements and general investigations: developmental plan for the drug into perspective and plans contingent on the outcome of the studies |
| Investigator's brochure |
| Chemistry, manufacturing, and control information |
| Pharmacology and toxicology information |
| Protocols (21 CFR 312.23(a) (6) |
| Previous human experience with the investigational drug: presented in an integrated summary report |
NOTE: From the Electronic Code of Federal Regulations (24).
Before describing in detail some key aspects of an IND submission for oncology products, the following caveats should be noted:
The PharmTox section of an IND contains an integrated summary and individual reports of nonclinical studies that support the IND.
It is expected that the sponsor provides significant understanding of the mechanism of action of the agent. However, in many circumstances, the exact mechanism of action for novel NMEs is unknown. In general, mechanism-of-action studies are initially done in in vitro models (e.g., tumor cell lines; ref. 8). Moreover, the DDOP expects that the sponsor will show antitumor effects in vivo in preclinical models. Sponsors should, in principle, show that at doses that are tolerated in nonclinical species, there is some effect on tumor growth, suggesting that there is a reasonable therapeutic index. Ideally, pharmacokinetic (see below) and pharmacodynamic studies should be done to better inform decisions about dose schedules and dose escalation schemes, whether in monotherapy and/or in combination with other established compounds.
Although pharmacokinetic studies are not required for IND submission, historically, the DDOP has found some pharmacokinetic information desirable, particularly when pharmacokinetic results are associated with antitumor effect and/or pharmacodynamic studies in nonclinical in vivo models. It is also desirable to have some information about absorption (particularly for oral small molecules), distribution, metabolism, and excretion in these animals, also known as ADME. Further, information is desirable about plasma protein-binding properties of the novel agent in several species, including humans. Pharmacokinetic information may assist the interspecies comparison of toxicity and extrapolation to humans. All this information may facilitate optimal dose escalation and dose schedule in the phase I trials. Pharmacokinetic information may suggest modifications in the intended dose, route, or schedule for the clinical trial. Nonclinical ADME data are generally complete by the time of submission of a new drug application (NDA).
Once an acceptable dose to administer to nonclinical species is obtained, a limited assessment of safety pharmacology should be conducted. The battery of tests includes the assessment of vital organ function: cardiovascular, respiratory, and central nervous systems, as part of the general toxicity studies. Of note, stand-alone safety pharmacology studies are not needed for phase I studies in patients with advanced cancer and could be done as part of the general PharmTox studies (8, 9). For more information, refer to ICH S7A guidance.
As mentioned earlier, the design of the definitive toxicity studies will depend on the intended clinical use. In this review, we will focus on the PharmTox data needed for patients with advanced malignancies. The main objectives for the PharmTox studies are outlined in Table 3. To this end, PharmTox studies should be done using schedules, formulations, durations, and routes comparable to those proposed for clinical studies.
Objectives of pharmacology and toxicology package at IND
| Estimate the safe starting dose for clinical studies |
| Assess toxic effects with respect to target organs (clinical pathology and histopathology), thus, potential organ toxicities can be monitored in human clinical studies |
| Assess reversibility of drug effect |
| Study varying dosing schedules and estimate the dose escalation scheme (in conjunction with the toxicokinetics) |
| Estimate the safe starting dose for clinical studies |
| Assess toxic effects with respect to target organs (clinical pathology and histopathology), thus, potential organ toxicities can be monitored in human clinical studies |
| Assess reversibility of drug effect |
| Study varying dosing schedules and estimate the dose escalation scheme (in conjunction with the toxicokinetics) |
NOTE: From S9 guidance.
In general, PharmTox studies are conducted in two mammalian species, rodent and nonrodent. The most typical species used are rat and dog. As mentioned earlier, this applies to small molecules as other biological agents may require other species including baboons, etc. Initial PharmTox studies are, in general, done in laboratories that are not necessarily certified for regulatory submissions. However, pivotal PharmTox studies, studies that will support the starting dose in phase 1 trials, should be conducted in accordance with Good Laboratory Practices (21CFR 58). The purpose of Good Laboratory Practices is to assure the integrity of the nonclinical safety data, such that an evaluation of the study quality and interpretation of the study results may be done with confidence (10).
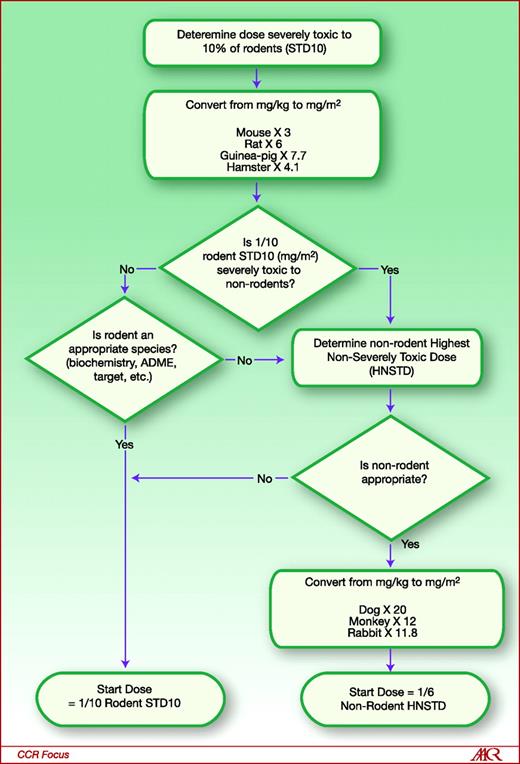
Most sponsors initially determine first the doses that are life-threatening and non-life-threatening in rodents. A severely toxic dose (i.e., dose that causes death or irreversible severe toxicity) in 10% of rodents (e.g., 1 out of 10 rats, STD 10) is generally obtained. Next, similar studies in nonrodent species using a range of doses that include STD10 (in mg/m 2 ) are conducted. The starting safe dose is based on the most sensitive species (see below and Fig. 1). At least one of the pivotal studies need to include clinical and pathology examination including gross and histopathology over a range of doses (from nontoxic to toxic). Moreover, pathological examination should be done off-therapy to show the reversibility of the findings. All these data will help determine which is the most sensitive species, whether there is a dose-response for adverse events, which organs are at risk for toxicity, which patients may be at-risk for toxicity, what needs to be monitored in the clinical trial, and what safe starting dose should be proposed. The issues that most concern FDA reviewers with respect to adverse events observed during nonclinical PharmTox studies are those that (1) are irreversible (particularly in crucial organs such as liver, kidneys, heart, eye), (2) are not amenable to monitoring (e.g., central nervous system), and (3) occur in a dose-independent manner.
General guide for starting dose selection for a cytotoxic agent in cancer patients. Reproduced courtesy of the U.S. FDA (22).
General guide for starting dose selection for a cytotoxic agent in cancer patients. Reproduced courtesy of the U.S. FDA (22).
As mentioned earlier, the safe starting dose is set on the basis of PharmTox studies in rodent and nonrodent species and the most sensitive species is chosen for safe starting dose determination. An example from a hypothetical case is presented for illustration. As shown in Fig. 1, the sponsor first determines the STD10 in rodents (11). In this example, the STD10 in the rat is 30 mg/kg of experimental drug. In order to compare the rodent dose and the nonrodent dose, the mg/kg dose must be converted to mg/m 2 . The conversion factor from mg/kg to mg/m 2 for the rat is ×6, leading to a dose of 180 mg/m 2 . The safe starting dose in humans is one tenth of the rat STD10. Thus, based on the rat PharmTox, the starting dose in humans would be 18 mg/m 2 . In parallel, studies in nonrodents (most typically dogs) are done. In this example, the highest nonseverely toxic dose (HNSTD), defined as the highest dose level that does not produce evidence of lethality, life-threatening toxicity, or irreversible findings, is 12 mg/kg. The conversion factor from mg/kg to mg/m 2 for the dog is ×20, yielding a dose of 240 mg/m 2 . The safe starting dose in humans is one sixth of the dog HNSTD; the safe starting dose in humans, on the basis of dog data, would thus be estimated at 40 mg/m 2 . The lowest of the two doses in mg/m 2 should be the actual starting dose in humans. In this particular case, the lowest dose is based on the rat, 18 mg/m 2 .
There are situations in which the starting dose may differ from the calculation above. There are some circumstances in which the nonrodent is the more appropriate species (i.e., rapid drug metabolism in the rat), then in that case, the nonrodent dose HNSTD should be used. In contrast, dogs are known to be poor predictors for toxicity for platinum-containing regimens (9, 12); thus, other nonrodent species should be used in these circumstances. Finally, the PharmTox reviewer at the FDA, on the basis of thorough review of the IND package, may disagree with these calculations, thereby reaching a different starting dose.
There are some caveats to the interpretation of nonclinical PharmTox. A retrospective review by Olson and colleagues evaluated the potential concordance between nonclinical and clinical toxicity of 150 compounds for all indications (including nononcology) that were tested in both nonclinical and clinical settings (13). The study found that when using both rodent and nonrodent toxicology information, there was a true concordance rate of 71% with human data. Instead, data from nonrodents alone or rodents alone have a 63% and 43% true concordance with human, respectively, suggesting that the use of both nonrodent and rodent species increases the true human prediction (14, 15). Unfortunately these data do not represent the whole spectrum of drugs at the preclinical level as the authors fail to analyze drugs that never reached the clinic because of unacceptable preclinical toxicity. Of note, most of the safety-related attrition (∼70%) occurs preclinically (16), and an estimated 20% of drug attrition is due to preclinical toxicity (17). Other preclinical prediction methods such as genomics and proteomics are being considered (16, 18). There are many reasons that may limit the extrapolation of nonclinical data to humans either qualitative (type of adverse event) or quantitative (dose that may cause adverse event or intensity of adverse event). These explanations include:
Despite all these limitations, although risks for humans cannot be eliminated, the focused intent of the IND-directed pharmacology and toxicology package is to anticipate, ameliorate, and/or avoid unacceptable adverse events. The usual practice of the DDOP for systemically administered drugs is to base a starting dose on a body surface area. Other approaches for determining a start dose may be considered as necessary. For instance, for intratumoral injections, it may be more appropriate to base the starting dose on concentration or milligrams injected.
In general, no additional preclinical combination PharmTox data are needed for combination therapy when both agents have been fully evaluated in humans (8, 9). However, when pharmacokinetic, metabolic, or pharmacodynamic interactions are expected, then PharmTox of the combination may be required. The other situation when combination PharmTox is needed is when drugs are packaged as a combination product. It is advisable to have a pre-IND consultation to discuss each particular case.
Other preclinical toxicity and/or safety studies that are part of an IND are genotoxicity, reproductive toxicity, and carcinogenicity. As mentioned earlier, the design of the definitive toxicities studies will depend on the intended clinical use. If the intended use is advanced and/or metastatic neoplasms, these studies are not required at the IND stage. However, if the intended use is in healthy volunteers, in the adjuvant setting, or chemoprevention, these studies should be submitted with the IND. Refer to the specific guidance as above (20).
All sections of the protocol to test a new compound in humans are important, however, the most contentious sections are: starting dose, dose escalation, population to be studied (inclusion and exclusion criteria), definition of dose-limiting toxicity, and monitoring of adverse events.
With respect to the safe starting dose, this should be based on PharmTox data (as discussed above and in Fig. 1). However, in INDs with prior clinical data (such as drugs that were tested in humans outside the United States), clinical safety information is considered more informative that nonclinical safety information.
Several potential dose escalation schemes can be proposed at the IND submission (9, 21). The most traditional one is the modified Fibonacci scheme in which there is a large initial dose escalation increase that becomes progressively smaller. Recently, other dose escalation designs have become more popular (2). In general, accelerated escalation designs involve doubling the dose in cohorts of one to three patients until a single grade 2 National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) adverse event occurs. At that time, dose escalation doubling stops and future cohorts have dose escalation increases that do not exceed 50% from previous cohorts. Although these designs are well accepted by the DDOP, there are at least three situations in which they may not be appropriate: (1) when the nonclinical dose-toxicity curve is steep, (2) when toxicities observed in nonclinical studies are unusual, irreversible, and/or dose-independent, and (3) for drugs intended for chronic dosing (21).
With respect to the population studied, in contrast to other therapeutic areas, healthy volunteers rarely participate in phase 1 oncology first-in-human studies. In oncology phase 1 trials, doses are escalated until a maximum tolerated dose is achieved (9, 21). Thus, the most typical population studied in a NME in oncology is advanced and/or metastatic solid tumor patients who have tumors that are refractory to standard therapies or for which no standard therapies exist, as the use of potentially subtherapeutic or overtly toxic experimental drugs in patients with chemosensitive tumors is considered unethical.
Finally, definition of dose-limiting toxicity (DLT) is one of the most contentious issues. A standard definition of DLT includes relevant toxicity not attributable to the disease or disease-related processes under investigation, which include: (1) hematological: neutropenia ≥ CTCAE grade 4 present for more than 4 days or associated with fever, grade 4 thrombocytopenia, or grade 3 thrombocytopenia and significant bleeding; (2) nonhematological: ≥CTCAE grade 3. In general, alopecia is excluded as part of the DLT. Moreover, ≥CTCAE grade 4 nausea and/or vomiting that lasts ≥24 hours despite supportive treatment is also considered DLT. Sponsors can propose exclusions to this definition (such as specific isolated laboratory changes without clinical consequences), and those can be discussed with DDOP at the IND stage. However, on the basis of specific adverse events observed in the PharmTox package, it is possible that DDOP will require a more conservative DLT definition such as specific ≥grade 2 adverse events that do not resolve to baseline or ≤grade 1 for 2 to 3 weeks. The definition of DLT and/or the approach to monitoring of adverse events may differ among different drugs on the basis of PharmTox findings or previous experience with drug or drug-class.
As soon as the IND is received at the FDA, the 30-day clock starts. If the FDA does not communicate with the sponsor by day 30, it is assumed that there are no problems and the protocol can proceed. However, in most cases (more than 90% of INDs), some issues need to be solved before initiation of the clinical trial. In general, the DDOP will contact the sponsor by fax before day 30 with two kinds of issues: (1) Deficiencies (requirements): sponsor is required to answer adequately (i.e., by protocol amendment) before day 30, otherwise, the protocol cannot proceed as written; or (2) Comments (recommendations): sponsor needs to answer but can elect not to follow FDA comments. If the sponsor agrees with amending the protocol and addressing the deficiencies, the protocol may proceed. However, if deficiencies are not satisfactorily addressed (i.e., unmonitorable toxicity or significant CMC issues), in general, an emergency teleconference is held between the DDOP and the sponsor before day 30. If deficiencies remain unsolved by day 30, the IND is placed on clinical hold and the protocol cannot start.
A clinical hold is an order issued by the FDA to the sponsor to delay a proposed clinical investigation or to suspend an ongoing investigation. When an ongoing study is placed on clinical hold, no new subjects may be recruited to the study; patients already in the study should be taken off therapy involving the investigational drug unless specifically permitted by FDA in the interest of patient safety.
The reasons for a clinical hold are: (1) Human subjects are or would be exposed to an unreasonable and significant risk of illness or injury (this is the most frequent reason for clinical hold); (2) The clinical investigators named in the IND are not qualified by reason of their scientific training and experience to conduct the investigation described in the IND; (3) The investigator brochure is misleading, erroneous, or materially incomplete; or (4) The IND does not contain sufficient information required under 21 CFR 312.23 to assess the risks to subjects of the proposed studies.
Once a clinical hold occurs, the sponsor needs to submit a letter to the FDA addressing all the deficiencies. If the FDA considers this response acceptable, then, the protocol may proceed.
To exempt a marketed drug product from the requirement to file an IND, five criteria need to be met [21 CFR 312.2 (b)]. The clinical study: (1) is not intended to support approval of a new indication or a significant change in the product labeling, (2) is not intended to support a significant change in advertising, (3) does not involve a route of administration or dosage level or use in a patient population or other factor that significantly increases the risk associated with the use of the drug product, (4) is conducted in compliance with the IRB and informed consent regulations, and (5) will not be used to promote unapproved indications. Again, an IND is not required in these circumstances. However, IRB approval and informed consent are still needed (21).
The main purpose of a pre-IND submission is to get feedback from the DDOP about the appropriateness of the initial development plan and to provide alternative pathways to initiating the phase 1 trial. The sponsor is encouraged to submit a pre-IND, particularly for unique products or unique questions that are important to address before all IND-required data are obtained.
In general, a pre-IND submission contains a study synopsis and preliminary nonclinical information. At the pre-IND stage, the DDOP will not conduct a detailed review of nonclinical data and will not provide protocol concurrence. These review issues are addressed at the IND stage.
As mentioned earlier the primary endpoint for phase I first-in-human trials is the assessment of safety and determination of recommended phase 2 dose. FDA expects that all sponsors obtain safety and efficacy data on the basis of acceptable standards. Data quality, discussed in a companion article in this issue of CCR Focus, is guided in general by FDA guidance and by the ICH and World Health Organization Good Clinical Practice guidelines (10).
In order to test a new drug product in the United States, the sponsor needs to submit an IND application. The package needs to contain sufficient information about the drug, investigators, and nonclinical pharmacology and toxicology data that would allow identification of a safe starting dose, protocol, and monitoring plan. The FDA has 30 days to respond to the sponsor. The response includes a list of deficiencies (concerns that need to be solved before initiation of trial) and/or recommendations (suggestions to improve the conduct of the trial). If the sponsor fails to address the deficiencies, the IND is placed on hold. Drug administration is halted until deficiencies are solved. Communication with the DDOP and pre-IND meetings provide the most efficient path to opening and conducting a phase I study (Table 4).
| Conduct pharmacodynamic, pharmacokinetic, and toxicity studies using the same schedule, duration, formulation, and route as proposed clinical trial. |
| Conduct rodent study that identifies life-threatening doses (STD 10). Then, conduct nonrodent study that confirms non-life threatening doses have been identified (HNSTD). |
| Provide histopathology in one of those studies. |
| Starting dose: in general is based on rodent information unless nonrodent is more sensitive or rodent is not an appropriate species. |
| IND clock 30 d: deficiencies need to be addressed. Otherwise, protocol(s) will be placed on clinical hold. |
| Pre-IND meetings may be very important for guidance and answers from FDA |
| Conduct pharmacodynamic, pharmacokinetic, and toxicity studies using the same schedule, duration, formulation, and route as proposed clinical trial. |
| Conduct rodent study that identifies life-threatening doses (STD 10). Then, conduct nonrodent study that confirms non-life threatening doses have been identified (HNSTD). |
| Provide histopathology in one of those studies. |
| Starting dose: in general is based on rodent information unless nonrodent is more sensitive or rodent is not an appropriate species. |
| IND clock 30 d: deficiencies need to be addressed. Otherwise, protocol(s) will be placed on clinical hold. |
| Pre-IND meetings may be very important for guidance and answers from FDA |
No potential conflicts of interest were disclosed.
I would like to acknowledge the Division of Drug Oncology Products, Office of Oncology Drug Products, FDA, and in particular Drs. John R. Johnson and Richard Pazdur for their mentoring and teaching regulatory medicine. I would also like to acknowledge Dr. Lourdes Villalba for helpful discussions about medical and regulatory issues.